Localization of orbitals¶
Example 1: Carbon dioxide¶
Textbooks and other sources, e.g. Wikipedia gives a Lewis structure of carbon dioxide with double bond from carbon to each oxygen. Is this the correct bonding picture ?
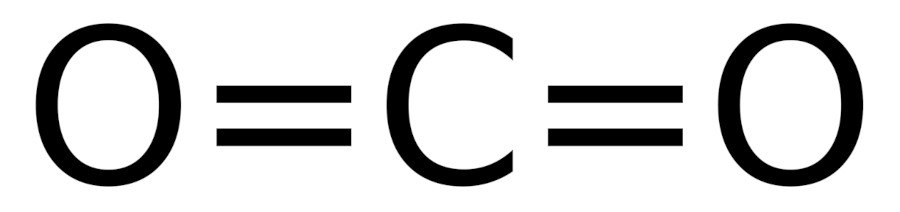
To investigate this, we shall carry out Pipek-Mezey localization [Pipek:Mezey] of the canonical molecular orbitals of carbon dioxide. We start from the molecular input CO2.xyz
3
C 0.0 0.0 0.0
O1 0.0 0.0 1.1647757354
O2 0.0 0.0 -1.1647757354
as well as the menu file CO2.inp
**DIRAC
.WAVE FUNCTION
**GENERAL
.ACMOUT
**HAMILTONIAN
.SPINFREE
.DFT
B3LYP
**WAVE FUNCTION
.SCF
*SCF
.CLOSED SHELL
12 10
**MOLECULE
*CENTERS
.NUCLEUS
O1 8.0
.NUCLEUS
O2 8.0
*BASIS
.DEFAULT
dyall.2zp
**END OF
Note that we distinguish the two oxygen atoms. Since we do not use the proper element name, the approprirate charges are given in the menu file using the NUCLEUS keyword. We run the SCF calculation using:
pam --inp=CO2 --mol=CO2.xyz
In preparation for localization we used the keyword .ACMOUT to dump \(C_1\) - coefficients to the checkpoint file. You can check this using:
h5dump -n CO2_CO2.h5 | grep C1
dataset /result/wavefunctions/scf/mobasis/eigenvalues_C1
dataset /result/wavefunctions/scf/mobasis/orbitals_C1
Now we are ready to localize the canonical MOs. We use the menu file loc.inp
**DIRAC
.ANALYZE
**GENERAL
.ACMOIN
**WAVE FUNCTION
.SCF
*SCF
.CLOSED SHELL
22
**ANALYZE
.LOCALIZE
*LOCALIZE
.THGRAD
1.0D-04
**MOLECULE
*SYMMETRY
.NOSYM
*CENTERS
.NUCLEUS
O1 8.0
.NUCLEUS
O2 8.0
*BASIS
.DEFAULT
dyall.2zp
**END OF
Note that we now have turned off symmetry (NOSYM) and ask to read the \(C_1\) coeffcients from the checkpoint file using the .ACMOIN keyword. We run localization using:
pam --inp=loc --mol=CO2.xyz --put "CO2_CO2.h5=DFACMO.h5"
Using grep we find:
$grep '*C' loc_CO2.out
*CENTERS
*C ITR DIFFA |GRAD| |KAPPA| H(nonD/D) e: +,-,0 H DIFFG DIFFQ HMIN
*C 0 0.00D+00 0.15D-01 0.00D+00 0.00115 158 54 8 FULL 0.00D+00 0.00D+00 -0.36D+00
*C 1 0.21D-01 0.86D-02 0.54D+01 0.00068 183 29 8 FULL -0.21D-01 -0.22D+01 -0.36D+00
*C 2 0.57D-02 0.78D-02 0.34D+00 0.00037 184 28 8 FULL -0.57D-02 -0.72D-02 -0.36D+00
*C 3 0.13D-02 0.53D-02 0.59D+00 0.00035 193 19 8 FULL -0.13D-02 -0.33D-02 -0.37D+00
*C 4 0.88D-03 0.99D-03 0.26D+00 0.00021 199 13 8 FULL -0.88D-03 -0.10D-02 -0.37D+00
*C 5 0.27D-04 0.32D-04 0.49D+00 0.00010 204 8 8 FULL -0.27D-04 -0.27D-04 -0.37D+00
*C 6 0.14D-07 0.28D-07 0.40D-01 0.00008 204 8 8 FULL -0.14D-07 -0.14D-07 -0.37D+00
*C 7 0.98D-01 0.81D-01 0.67D+00 0.00143 203 9 8 FULL -0.98D-01 -0.17D+00 -0.26D+00
*C 8 0.54D-01 0.89D-01 0.30D+00 0.00130 201 11 8 FULL -0.54D-01 -0.64D-01 -0.18D+00
*C 9 0.23D-01 0.52D-01 0.16D+00 0.00116 201 11 8 FULL -0.23D-01 -0.25D-01 -0.18D+00
*C 10 0.78D-02 0.65D-02 0.17D+00 0.00103 203 9 8 FULL -0.78D-02 -0.86D-02 -0.18D+00
*C 11 0.42D-04 0.50D-02 0.40D+01 0.00104 205 7 8 FULL -0.42D-04 -0.10D-03 -0.18D+00
*C 12 0.68D-04 0.73D-04 0.59D+00 0.00104 208 4 8 FULL -0.68D-04 -0.70D-04 -0.18D+00
*C 13 0.64D-06 0.73D-05 0.39D+00 0.00103 208 4 8 FULL -0.64D-06 -0.16D-05 -0.18D+00
*C 14 0.39D-06 0.74D-06 0.13D+00 0.00104 208 4 8 FULL -0.39D-06 -0.41D-06 -0.18D+00
*C 15 0.28D-08 0.41D-08 0.10D-01 0.00103 208 4 8 FULL -0.28D-08 -0.28D-08 -0.18D+00
*C 16 0.25D-01 0.73D-02 0.40D+01 0.00101 206 6 8 FULL -0.25D-01 -0.16D+01 -0.18D+00
*C 17 0.42D-03 0.12D-03 0.62D-01 0.00100 209 3 8 FULL -0.42D-03 -0.42D-03 -0.18D+00
*C 18 0.13D-06 0.10D-07 0.50D-02 0.00100 209 3 8 FULL -0.13D-06 -0.13D-06 -0.18D+00
*C 19 0.58D-05 0.32D-04 0.10D+01 0.00099 210 2 8 FULL -0.58D-05 -0.28D-04 -0.18D+00
*C 20 0.11D-01 0.41D-01 0.25D+00 0.00100 202 16 2 FULL -0.11D-01 -0.11D-01 -0.14D+00
*C 21 0.79D-01 0.70D-02 0.82D+00 0.00033 212 6 2 FULL -0.79D-01 -0.16D+00 -0.11D-03
*C 22 0.27D-03 0.28D-04 0.28D+00 0.00033 212 6 2 FULL -0.27D-03 -0.27D-03 -0.18D-08
*C 23 0.14D-06 0.25D-06 0.72D-01 0.00033 212 0 8 FULL -0.14D-06 -0.14D-06 0.00D+00
Let us now have a look at the localized orbitals. The Pipek-Mezey criterion minimizes the number of centers spanned by each MO. We accordingly perform a Mulliken population analysis, asking only for the distribution of density amongst the three atoms. The menu file atompop.inp reads
**DIRAC
.ANALYZE
**WAVE FUNCTION
.SCF
*SCF
.CLOSED SHELL
22
**ANALYZE
.MULPOP
*MULPOP
.LABEL
ATOM
**MOLECULE
*SYMMETRY
.NOSYM
*CENTERS
.NUCLEUS
O1 8.0
.NUCLEUS
O2 8.0
*BASIS
.DEFAULT
dyall.2zp
**END OF
We run the calculation using:
pam --inp=atompop --mol=CO2.xyz --put "loc_CO2.h5=CHECKPOINT.h5"
In the output we find:
**************************************************************************
********************** Mulliken population analysis **********************
**************************************************************************
Fermion ircop E1
----------------
* Electronic eigenvalue no. 1: -19.228904376854 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L O1 1
--------------------------------------
alpha 1.0000 | 1.0086
beta 0.0000 | 0.0000
* Electronic eigenvalue no. 2: -19.228897406456 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L O2 1
--------------------------------------
alpha 1.0000 | 1.0086
beta 0.0000 | 0.0000
* Electronic eigenvalue no. 3: -10.375624527418 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L C 1
--------------------------------------
alpha 0.9997 | 1.0010
beta 0.0003 | 0.0000
* Electronic eigenvalue no. 4: -1.1701747687870 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L O2 1
--------------------------------------
alpha 0.9995 | 0.9988
beta 0.0005 | 0.0000
* Electronic eigenvalue no. 5: -1.1313373272171 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L C 1 L O1 1
-----------------------------------------------------
alpha 1.0000 | 0.4569 0.5497
beta 0.0000 | 0.0000 0.0000
* Electronic eigenvalue no. 6: -0.5710187998784 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L C 1 L O2 1
-----------------------------------------------------
alpha 1.0000 | 0.4569 0.5497
beta 0.0000 | 0.0000 0.0000
* Electronic eigenvalue no. 7: -0.5281308034633 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L O1 1
--------------------------------------
alpha 0.9995 | 0.9988
beta 0.0005 | 0.0000
* Electronic eigenvalue no. 8: -0.5229733977076 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L C 1 L O1 1
-----------------------------------------------------
alpha 1.0000 | 0.2282 0.7706
beta 0.0000 | 0.0000 0.0000
* Electronic eigenvalue no. 9: -0.5229733976932 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L C 1 L O1 1
-----------------------------------------------------
alpha 1.0000 | 0.2282 0.7706
beta 0.0000 | 0.0000 0.0000
* Electronic eigenvalue no. 10: -0.3795760457099 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L C 1 L O2 1
-----------------------------------------------------
alpha 1.0000 | 0.2282 0.7706
beta 0.0000 | 0.0000 0.0000
* Electronic eigenvalue no. 11: -0.3795760457064 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L C 1 L O2 1
-----------------------------------------------------
alpha 1.0000 | 0.2282 0.7706
beta 0.0000 | 0.0000 0.0000
*** Total gross population ***
Gross Total | L C 1 L O1 1 L O2 1
--------------------------------------------------------------------
total 22.00000 | 5.62427 8.18536 8.18536
Simple observation shows that there are three and not two localized orbitals connecting carbon to each oxygen atom, suggesting triple and not double bonds. Before exploring this further, it is important to note that the orbitals energies given in the output are those of the canonical MOs. Localized MOs do not have orbital energies. However, to establish some energetic ordering we can calculate approximate orbital energies as expectation values of each localized orbital with respect to the Kohn-Sham matrix (for HF calculations one would use the Fock matrix).
Since we can not use molecular symmetry with the localized orbitals we first have to reconstruct the Kohn-Sham matrix for the \(C_1\) case. We prepare the input file scf0.inp
**DIRAC
.WAVE FUNCTION
**HAMILTONIAN
.SPINFREE
.DFT
B3LYP
**WAVE FUNCTION
.SCF
*SCF
.CLOSED SHELL
22
.MAXITR
0
**MOLECULE
*SYMMETRY
.NOSYM
*CENTERS
.NUCLEUS
O1 8.0
.NUCLEUS
O2 8.0
*BASIS
.DEFAULT
dyall.2zp
**END OF
Note that we set the number of SCF iterations to zero such that we only build the initial Kohn–Sham matrix with the input coefficients (no diagonalization). Next we run:
pam --inp=scf0 --mol=CO2.xyz --put "loc_CO2.h5=CHECKPOINT.h5" --get DFFCKT
where we recover the total Kohn-Sham matrix in the unformatted file DFFCKT. To see that our procedure actually works let us first calculate the expectation value of this matrix with respect to the original canonical MOs. Using the input file exp0.inp
**DIRAC
.PROPERTIES
**GENERAL
.ACMOIN
**WAVE FUNCTION
.SCF
*SCF
.CLOSED SHELL
22
**PROPERTIES
*EXPECTATION VALUE
.ORBANA
.OPERATOR
'Kohn-Sham'
FOCKMAT
DFFCKT
FACTOR
1.0D0
**MOLECULE
*SYMMETRY
.NOSYM
*CENTERS
.NUCLEUS
O1 8.0
.NUCLEUS
O2 8.0
*BASIS
.DEFAULT
dyall.2zp
**END OF
we run:
pam --inp=expC --mol=CO2.xyz --put "CO2_CO2.h5=DFACMO.h5 DFFCKT"
Note that we use the keyword .ACMOIN since want to use the \(C_1\) coefficients from the original DFT calculation with symmetry. At the end of the generated output file we then find back the orbital energies that are found in the Mulliken population analysis above:
Expectation value for individual orbitals
-----------------------------------------
Matrix element Occ.
E1 1 -19.22890437800 2.0000 -38.45780875600
E1 2 -19.22889740759 2.0000 -38.45779481518
E1 3 -10.37562453182 2.0000 -20.75124906363
E1 4 -1.170174770392 2.0000 -2.340349540785
E1 5 -1.131337328982 2.0000 -2.262674657965
E1 6 -0.571018800591 2.0000 -1.142037601183
E1 7 -0.528130804300 2.0000 -1.056261608600
E1 8 -0.522973398844 2.0000 -1.045946797688
E1 9 -0.522973398844 2.0000 -1.045946797688
E1 10 -0.379576046966 2.0000 -0.759152093933
E1 11 -0.379576046966 2.0000 -0.759152093933
-------------------------------------------------------------------------------------
Total : -108.0783738266 a.u. s0 = F t0 = F
-------------------------------------------------------------------------------------
s0 = T : Expectation value zero by point group symmetry.
t0 = T : Expectation value zero by time reversal symmetry.
Now we do the same with the localized orbitals and combine this with a mode detailed Mulliken population analysis. We set up the input file exp.inp
**DIRAC
.PROPERTIES
.ANALYZE
**WAVE FUNCTION
.SCF
*SCF
.CLOSED SHELL
22
**ANALYZE
.MULPOP
**PROPERTIES
*EXPECTATION VALUE
.ORBANA
.OPERATOR
'Kohn-Sham'
FOCKMAT
DFFCKT
FACTOR
1.0D0
**MOLECULE
*SYMMETRY
.NOSYM
*CENTERS
.NUCLEUS
O1 8.0
.NUCLEUS
O2 8.0
*BASIS
.DEFAULT
dyall.2zp
**END OF
and run:
pam --inp=exp --mol=CO2.xyz --put "loc_CO2.h5=CHECKPOINT.h5 DFFCKT"
In the new Mulliken population analysis we still have the orbital energies of the canonical orbitals, but in the output below I have replaced these with the expectation values for the localized orbitals and also reordered the orbitals on energy. I have kept the original numbering, though. We now have:
* Electronic eigenvalue no. 7: -17.16989574591 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A O1 s L A O1 pz
-----------------------------------------------------
alpha 0.9995 | 0.9858 0.0130
beta 0.0005 | 0.0000 0.0000
* Electronic eigenvalue no. 4: -17.14806997582 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A O2 s L A O2 pz
-----------------------------------------------------
alpha 0.9995 | 0.9856 0.0132
beta 0.0005 | 0.0000 0.0000
* Electronic eigenvalue no. 3: -10.33132604930 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A C s
--------------------------------------
alpha 0.9997 | 1.0010
beta 0.0003 | 0.0000
* Electronic eigenvalue no. 2: -2.821559678558 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A C s L A O2 s L A O2 pz
--------------------------------------------------------------------
alpha 1.0000 | -0.0108 0.8393 0.1692
beta 0.0000 | -0.0000 0.0000 0.0000
* Electronic eigenvalue no. 1: -2.799733777067 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A C s L A O1 s L A O1 pz
--------------------------------------------------------------------
alpha 1.0000 | -0.0108 0.8391 0.1694
beta 0.0000 | -0.0000 0.0000 0.0000
* Electronic eigenvalue no. 5: -0.981751468005 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A C s L A C pz L A C dzz L A O1 s L A O1 pz L A O1 dzz
-----------------------------------------------------------------------------------------------------------------
alpha 1.0000 | 0.2676 0.1794 0.0284 0.0792 0.4654 0.0133
beta 0.0000 | 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000
* Electronic eigenvalue no. 6: -0.981751327023 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A C s L A C pz L A C dzz L A O2 s L A O2 pz L A O2 dzz
-----------------------------------------------------------------------------------------------------------------
alpha 1.0000 | 0.2676 0.1794 0.0284 0.0792 0.4654 0.0133
beta 0.0000 | 0.0000 0.0000 0.0000 0.0000 0.0000 0.0000
* Electronic eigenvalue no. 8: -0.451274722906 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A C py L A C dyz L A O1 py
--------------------------------------------------------------------
alpha 1.0000 | 0.2034 0.0248 0.7677
beta 0.0000 | 0.0000 0.0000 0.0000
* Electronic eigenvalue no. 9: --0.451274722906 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A C px L A C dxz L A O1 px
--------------------------------------------------------------------
alpha 1.0000 | 0.2034 0.0248 0.7677
beta 0.0000 | 0.0000 0.0000 0.0000
* Electronic eigenvalue no. 10: -0.451274722904 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A C px L A C dxz L A O2 px
--------------------------------------------------------------------
alpha 1.0000 | 0.2034 0.0248 0.7677
beta 0.0000 | 0.0000 0.0000 0.0000
* Electronic eigenvalue no. 11: -0.451274722904 (Occupation : f = 1.0000)
============================================================================================
* Gross populations greater than 0.01000
Gross Total | L A C py L A C dyz L A O2 py
--------------------------------------------------------------------
alpha 1.0000 | 0.2034 0.0248 0.7677
beta 0.0000 | 0.0000 0.0000 0.0000
We can see that the first three orbitals are clearly the \(1s\) orbitals of the three atoms of the molecule. Then comes two orbitals dominated by s-orbitals on the oxygens. Finally comes \(\sigma\)-orbitals from carbon to each oxygen atom (\(\langle\varepsilon\rangle=-0.982\ E_h\)), followed by pairs of \(\pi\)-orbitals to each oxygen (\(\langle\varepsilon\rangle=-0.451\ E_h\))